
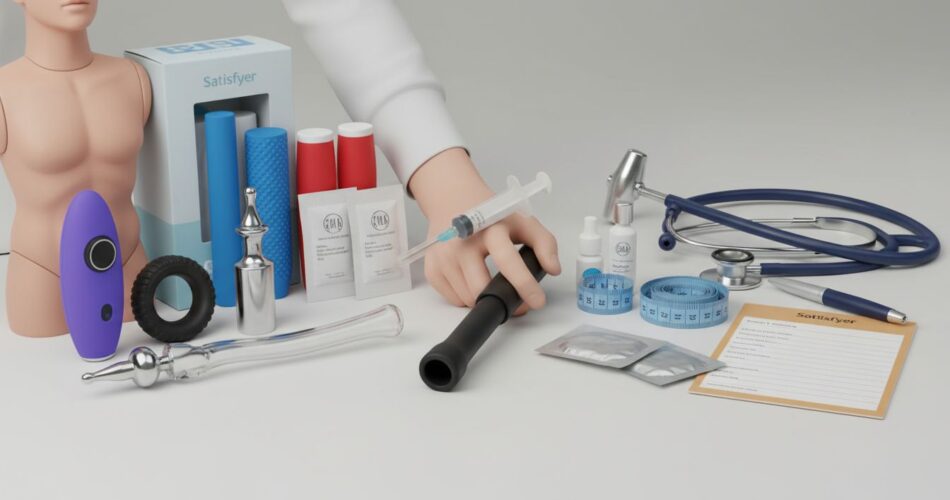
En los últimos años hemos visto una transformación notable: lo que antes se veía como entretenimiento y ocio se reivindica ahora como parte de la atención sanitaria. Empresas del sector conocido como sex‑tech están reposicionando vibradores, wearables y dispositivos pélvicos como «dispositivos terapéuticos», buscando ensayos clínicos, certificaciones regulatorias y vías de reembolso para ganar legitimidad médica.
Este movimiento no es solo de marketing. Hay cambios en el marco regulatorio, evidencia clínica emergente (aunque a menudo limitada) y una demanda palpable por parte de pacientes y profesionales. A medida que surgieron códigos regulatorios como el product‑code KXQ de la FDA y ensayos sobre dispositivos como Crescendo o FemPulse, la línea entre lo lúdico y lo clínico se vuelve más difusa.
Del ocio a la salud: una tendencia en marcha
El sector sex‑tech experimentó una migración conceptual entre 2022 y 2026: fabricantes están redefiniendo vibradores y wearables como herramientas de salud sexual y rehabilitación. Esta evolución responde tanto a intereses comerciales (ampliar mercado) como a la demanda clínica de intervenciones no farmacológicas.
Importantes actores del mercado han comunicado su intención de financiar ensayos clínicos y buscar autorizaciones regulatorias. El objetivo declarado es doble: demostrar eficacia y abrir rutas de reembolso que legitimen el uso médico frente a usuarios y pagadores.
La conversión de productos de ocio a dispositivos médicos también implica cambios en el diseño, la documentación y la relación con profesionales sanitarios: desde instrucciones para uso terapéutico hasta colaboraciones con fisioterapeutas y urólogos.
Tamaño del mercado y motivaciones comerciales
El mercado global de sex‑toys se estimó en alrededor de USD 32.7, 33 mil millones en 2022, con proyecciones de crecimiento de dos dígitos a mediano plazo según informes sectoriales (Grand View Research, Global Market Insights, 2024, 2026). Ese volumen explica por qué muchas empresas buscan rutas clínicas y de reembolso.
La posibilidad de acceder a fondos FSA/HSA, cubrir costes mediante seguros privados o integrarse en programas postparto o de rehabilitación aumenta el valor unitario y la sostenibilidad comercial. Marcas como MysteryVibe han hecho públicas sus estrategias sobre FSA/HSA y registro FDA.
Pero el interés comercial genera también tensiones: la presión por resultados positivos y la financiación industrial pueden afectar la independencia de la investigación, lo que obliga a una lectura crítica de los datos disponibles.
Vías regulatorias: el caso del product‑code FDA KXQ
En EE. UU. existe un product‑code específico en la FDA para «Vibrator For Therapeutic Use, Genital» (KXQ; 21 C.F.R. 884.5960). Este código ha servido como referencia para fabricantes que desean registrar dispositivos vibratorios con un propósito terapéutico.
La ruta regulatoria puede incluir registro/listado y, en algunos casos, 510(k) si el dispositivo reclama una indicación médica clara. Es importante distinguir entre estar registrado en la base de la FDA y haber obtenido un cleared/approved 510(k): no todos los productos con registro tienen aprobación premarket para indicaciones específicas.
La guía Device Advice de la FDA y las interacciones con agencias regulatorias como Health Canada o autoridades CE también han favorecido que plataformas como EMSELLA amplíen autorizaciones para indicaciones relacionadas con incontinencia y potencialmente función sexual.
Evidencia clínica: hallazgos, límites y estudios clave
La literatura sugiere señales prometedoras. Por ejemplo, un estudio multicéntrico preliminar publicado en abril de 2022 sobre el dispositivo Crescendo (MysteryVibe) reportó una mejora estadísticamente significativa en la subescala de dolor del FSFI (media pre 0.40 → post 2.32; p=0.02) tras 12 semanas.
Hay además series de casos y presentaciones en congresos (ESSM, Journal of Sexual Medicine) sobre wearables como Tenuto y otros dispositivos para disfunción eréctil o estimulación peneana, que muestran mejoras en ciertas cohortes. Productos como Elvie Trainer cuentan con uso clínico establecido para entrenamiento de Kegels y rehabilitación del suelo pélvico.
No obstante, las revisiones sistemáticas (p. ej. Sexual Medicine Reviews, ene 2023) concluyen que la evidencia es todavía limitada, heterogénea y en muchos casos patrocinada por la industria. Son recurrentes las series pequeñas, abstracts y falta de ECA amplios y de financiación independiente, lo que obliga a prudencia interpretativa.
Modelos tecnológicos y aplicaciones clínicas: Elvie, EMSELLA, Tenuto, FemPulse y otros
Los dispositivos se diversifican: desde trainers de suelo pélvico con biofeedback (Elvie Trainer) hasta plataformas de neuromodulación no invasiva (EMSELLA) y anillos o wearables para estimulación genital (Tenuto, Crescendo). Cada categoría plantea usos clínicos distintos.
Elvie Trainer se comercializa como un medical device usado en programas de rehabilitación postparto y en la práctica de fisioterapia pélvica, aportando datos cuantitativos para el entrenamiento de Kegels. EMSELLA, por su parte, ha ampliado autorizaciones regulatorias para indicaciones de incontinencia y anuncia extensiones para mejorar la función sexual.
FemPulse ejemplifica la búsqueda de trayectorias regulatorias formales: en diciembre de 2024 obtuvo autorización para iniciar un ensayo pivotal en sobre‑actividad vesical. Estos desarrollos muestran que la femtech no solo investiga, sino que está entrando en fases regulatorias formales.
Reembolso, aceptación clínica y retos éticos
Algunas marcas han conseguido elegibilidad FSA/HSA y promueven inclusión en rutas de pago, pero la cobertura pública y aseguradora sigue fragmentaria. El reembolso requiere evidencia robusta, códigos específicos y procesos burocráticos que no siempre favorecen productos emergentes.
Encuestas clínicas en poblaciones uro‑ginecológicas (2024, 2025) indican que una proporción significativa de pacientes y muchos clínicos consideran aceptable que se recomienden vibradores o programas de ejercicios para mejorar función sexual y síntomas del suelo pélvico. Reportes de pacientes muestran mejoras en dolor, confianza y síntomas urinarios.
Sin embargo, existen consideraciones éticas y prácticas: conflictos de interés en estudios patrocinados, necesidad de formación clínica para prescribir/usar correctamente estos dispositivos, y el riesgo de medicalizar productos que también tienen un componente lúdico. Asociaciones profesionales (p. ej. APTA, marzo 2024) han observado que algunos dilatadores y equipos íntimos se tratan como dispositivos médicos de clase II, lo que tiene implicaciones para prescripción y reembolso.
Riesgos, limitaciones y la necesidad de más investigación
Los riesgos reportados en la literatura suelen ser bajos, pero la calidad metodológica de muchos estudios es limitada: tamaños muestrales reducidos, seguimiento corto, ausencia de controles aleatorizados y dependencia de fuentes financiadas por la industria.
Para consolidar la legitimidad médica se requieren ECA bien diseñados, registros independientes, transparencia sobre conflictos de interés y estudios de costo‑efectividad que permitan a los pagadores valorar reembolso. La comunidad científica reclama investigaciones neutrales y replicaciones.
Mientras tanto, la práctica clínica puede apoyarse en la evidencia disponible, siempre informando a pacientes sobre beneficios potenciales, incertidumbres y alternativas, y documentando resultados en contextos reales para construir evidencia de implementación.
La migración de lo lúdico a lo clínico en el terreno de los dispositivos íntimos es real y multifacética: combina interés comercial, avances regulatorios y evidencia prometedora, pero incompleta. Los desarrollos como el product‑code KXQ, los estudios sobre Crescendo y los ensayos en curso (FemPulse, Tenuto, etc.) muestran un sector que busca rigor y legitimidad clínica.
Para que estos dispositivos sean plenamente aceptados en la práctica médica se necesitan más ensayos independientes, claridad regulatoria y rutas de reembolso sostenibles. Pacientes y clínicos son cada vez más receptivos, pero la medicina basada en evidencia exige prudencia: hay que balancear innovación, seguridad y transparencia.